
Assessment of theoretical prediction of the NMR shielding tensor of ¹⁹⁵PtClxBr6–x²⁻ complexes by DFT calculations: experimental and computational results
- Penka Fowe, Emmanuel Chemistry Department, University of Fribourg, Switzerland
- Belser, Peter Chemistry Department, University of Fribourg, Switzerland
- Daul, Claude Chemistry Department, University of Fribourg, Switzerland
- Chermette, Henry Laboratoire de Chimie Physique Théorique, Université Claude Bernard Lyon-1, Villeurbanne, France
-
17.04.2005
Published in:
- Physical Chemistry Chemical Physics. - 2005, vol. 7(8), p. 1732-1738
English
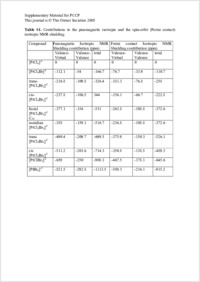
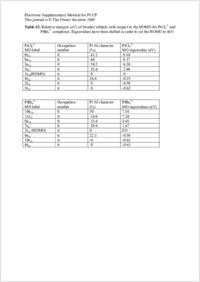
In the present work, the ZORA spin–orbit Hamiltonian, in conjunction with the gauge including orbital (GIAO) method based on DFT theory has been used to calculate ¹⁹⁵Pt chemical shift of ¹⁹⁵PtClxBr6–x²⁻ complexes. Excellent agreement with experiments has been obtained for calculations bearing on optimized geometries and all electrons triple zeta + polarization (TZP) STO basis sets: the relative error with respect to experiment amounts to <1.5%. It is found that the Pt chemical shift is dominated by the paramagnetic and the spin orbit contribution, whereas the diamagnetic term remains negligible. The influence of the quality of the basis sets has been studied and found to be small, provided a basis set like TZP is used. Several calculations have been performed in order to establish the sensitivity of the chemical shift to a variation in the bond lengths. A strong dependence has been found, with an increase of the chemical shift amounting to 150 ppm pm⁻¹ for a distance decrease. Large sensitivity to the solvat on, leading to changes in the structure, is then expected. Different tests using conductor-like screening models have been performed in order to establish the sensitivity of the chemical shift to solvation. It has been observed that the changes in the geometry are more important than charge transfers. Finally, the sensitivity of the system to the exchange–correlation functional is found rather weak, at least among the GGA functionals.
- Faculty
- Faculté des sciences et de médecine
- Department
- Département de Chimie
- Language
-
- English
- Classification
- Chemistry
- Other electronic version
- License
-
License undefined
- Identifiers
-
- RERO DOC 4794
- DOI 10.1039/b500574d
- Persistent URL
- https://folia.unifr.ch/unifr/documents/299715
Other files
Statistics
Document views: 306
File downloads:
- Texte intégral: 435
- Texte intégral: 182
- Texte intégral: 185